Maladie de la vache folle : combien d'humains seront contaminés ?
Combien de victimes fera la variante humaine de la maladie de la vache folle ? La question est posée depuis de nombreuses années et chaque nouvelle étude y apporte une réponse différente. La tendance générale, confirmée par une étude publiée en avril 2003, est de réviser les estimations à la baisse. Mais de nombreuses questions restent en suspens.
Philippe Dorison - Publié le
40 nouveaux cas de 2003 à 2008 ?
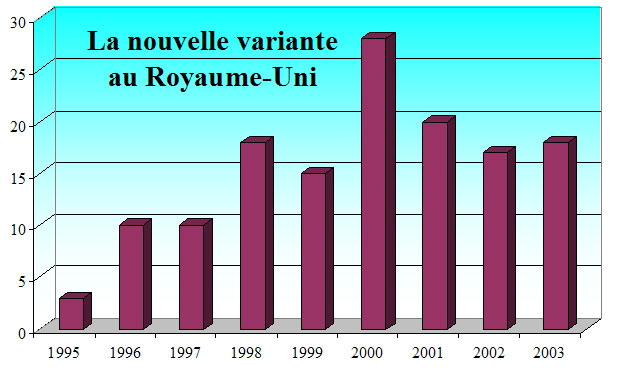
L’étude publiée en avril 2003 par le département d’épidémiologie des maladies infectieuses de l’Imperial College est beaucoup moins pessimiste que celle réalisée par la même équipe en 2000. Alors que le nombre maximal de nouveaux cas d’ici 2040 y était évalué à 136 000, il est revu à 2 600. La plus basse valeur de la dernière fourchette de prédiction (elle aussi fournie avec une probabilité de 95%) passe de 70 à 10.
Concernant les prévisions à court terme, le chiffre le plus probable retenu est de 40 nouveaux cas de 2003 à 2008, l’intervalle retenu s’étendant de 9 à 540 malades. L’évolution de ces prédictions s’explique principalement par le nombre de cas diagnostiqués qui ont pu être intégrés dans chaque étude (121 en 2003 contre 53 en 2000) et par la baisse constatée du nombre de nouveaux cas apparus au Royaume-Uni entre 2000 et 2002.
Les épidémiologistes de l’Imperial College envisagent donc que seuls 20 cas du nouveau variant de la maladie de Creutzfelt-Jakob pourraient se déclarer entre 2004 et 2005. Entre ces deux études, d’autres équipes avaient publié des travaux sur le même sujet. Leurs conclusions s’inscrivaient dans cette tendance régulière à la baisse que l’on peut aujourd’hui constater sur un intervalle de trois ans. Chacune de ces publications s’appuyait sur un nombre croissant de cas constatés (84 et 97 pour les études datées de 2001).
Au fil des années, les scientifiques disposent d'un nombre croissant de données pour affiner leur travail et l’approche de la maladie devient plus précise. Mais la fourchette de prévision reste large car ces différentes études doivent se baser sur un certain nombre d’hypothèses qui ne sont pas systématiquement validées par des résultats scientifiques incontestables.
Les principales études publiées entre 2000 et 2003
- Updated projections of future vCJD deaths in the UK
(BMC Infectious Diseases 2003, 3:4)
Azra C Ghani, Christl A Donnelly, Neil M Ferguson and Roy M Anderson Department of Infectious Disease Epidemiology, Faculty of Medicine, Imperial College of Science, Technology and Medicine, Norfolk Place, London W2 1PG, UK
- Deaths from variant Creutzfeldt-Jakob disease in the UK.
(Lancet. 2003 Mar 1; 361(9359): 751-2 )
Andrews NJ, Farrington CP, Ward HJ, Cousens SN, Smith PG, Molesworth AM, Knight RS, Ironside JW, Will RG. Public Health Laboratory Service, Communicable Disease Surveillance Centre, London, UK.
- Predictability of the UK variant Creutzfeldt-Jakob disease epidemic.
(Science. 2001 Nov 23; 294(5547): 1729-31.)
Aignaux JN, Cousens SN, Smith PG.
- Estimation of epidemic size and incubation time based on age characteristics of vCJD in the United Kingdom. (Science. 2001 Nov 23;294(5547):1663-4.)
- Alain-Jacques Valleron, Unité Inserm 444, CHU Saint-Antoine, Université Pierre et Marie Curie, Paris ;
- Robert Will, Unité nationale de surveillance de la maladie de Creutzfeldt-Jakob, Édimbourg, Royaume-Uni ;
- Jean-Yves Cesbron, UFR de médecine de Grenoble, Université Joseph Fourier, Grenoble.
- Predicted vCJD mortality in Great Britain. (Nature. 2000 Aug 10; 406(6796): 583-4)
Ghani AC, Ferguson NM, Donnelly CA, Anderson RM.
Certitudes et suppositions
Si les chercheurs s’accordent sur le nombre de bovins en incubation de l’ESB consommés au Royaume-Uni de 1980 à 1989 (entre 500 000 et un million), cela ne permet pas de connaître le nombre d’humains que ces animaux ont réellement contaminé.
En effet, la quantité de nourriture infectée susceptible d'induire la maladie chez l'homme est inconnue et le restera probablement. Il est évident qu'aucune expérience ne peut être faite à ce sujet chez l'humain et les modèles animaux ne permettent pas d'aboutir à des conclusions probantes.
Cette question rejoint celle de la « barrière d’espèce » que la maladie a été capable de traverser pour se transmettre des bovins aux humains. Les dernières projections sur l’étendue de l’épidémie humaine laissent supposer que la barrière d'espèce est plus difficile à franchir qu’on aurait pu le craindre il y a quelques années, sans qu’il soit toutefois possible de le mesurer avec précision.
Dans les points positifs, il faut aussi noter que la décroissance de l'épidémie d'ESB au Royaume-Uni et l'adoption de nombreuses mesures de protection de la santé publique laissent espérer que quasiment aucune contamination ne s'est produite après 1989.
La durée d'incubation, objet de toutes les hypothèses
Pendant la période d'incubation, la maladie est silencieuse et la personne infectée ne présente aucun symptôme.
Les derniers travaux de l’équipe de l’Imperial College considèrent comme probable que cette période se situerait entre 12,6 et 16,7 années, sans toutefois exclure qu’elle pourrait aller jusqu’à 35 ans. Une durée voisine (16 ans et 7 mois en moyenne, dans une fourchette de 12 à 23 ans) avait été retenue par l’équipe pilotée par Alain-Jacques Valleron pour l’Inserm, dans son étude publiée en 2001.
Ce paramètre est probablement celui qui conditionne le plus fortement l’évolution future de l’épidémie. Ainsi, en envisageant une durée d’incubation moyenne supérieure à 60 ans, l’équipe de l’Imperial College était parvenue en 2000 à une nombre maximal de cas qui pouvait atteindre 136 000. Ce chiffre descendait à 3 000 si la période d’incubation ne dépassait pas 30 ans.
La seule comparaison avec d’autres maladies semblables concerne le Kuru, une encéphalopathie qui s’est développée en Papouasie Nouvelle Guinée dans les années 50 et qui, elle, n’avait pas à franchir la barrière d’espèce. Certains cas sont apparus jusqu’à 40 ans après la contamination, pour une durée moyenne d’incubation qui s’établit entre 10 et 13 ans, 90% des cas s’étant déclenchés moins de 27 ans après l’infection. Une aussi longue période d’incubation est cohérente avec ce qui est observé chez les bovins, où elle atteint couramment cinq ans et plus, une durée extrêmement longue pour une maladie animale.
Quels moyens de dépistage ?
Aucun moyen fiable de dépistage ne permet de détecter le prion pathologique dans un organisme vivant.
Quels enjeux ?
La possibilité de détecter la trace du prion, l'agent infectieux, chez un animal vivant et apparemment sain serait l'un des meilleurs moyens d'éradiquer la maladie en abattant sélectivement les animaux touchés.
Côté humain, le bénéfice serait moins net tant que la médecine ne dispose d'aucun moyen de guérir ni même de freiner la maladie. Mais dès que des stratégies thérapeutiques commenceront à se mettre en place, l'enjeu du dépistage deviendra crucial.
Pourtant, avant même de savoir soigner cette maladie, un moyen de diagnostic fiable et précoce permettrait de prendre des précautions particulières lorsqu'un patient en incubation doit subir une intervention chirurgicale. En effet, les procédures de désinfection couramment utilisées pour les instruments sont inefficaces dans le cas d'une contamination par le prion pathologique.
Quelle recherche ?
Si elle est aujourd'hui très active, la recherche sur les maladies à prions est pourtant presque ''débutante'' : avant 1996, elle n'occupait que très peu d'équipes et elle a surtout été stimulée par les pouvoirs publics depuis la crise de l'automne 2000.
Dans le domaine du dépistage, une voie prometteuse a été ouverte par une équipe suisse (Claudio Soto et Gabriela Saborio, des laboratoires Serono) qui a mis au point une technique d'amplification des prions grâce aux ultra-sons. Les plaques de protéines infectieuses, agrégées entre elles, sont démantelées par ce traitement et se multiplient jusqu'à être suffisamment nombreuses pour devenir détectables. On peut ainsi espérer repérer l'infection chez des bovins en début d'incubation, lorsque seules des parties comme l'iléon (extrémité de l'intestin) sont infectées.
D'autres organes extérieurs au système nerveux central, comme le thymus et les amygdales pourraient aussi être examinés, permettant de repérer l'infection plus tôt et avec une meilleure fiabilité, y compris chez les humains. Des travaux publiés en février 2003 par une équipe italienne suggèrent même que le prion pourrait être détecté au niveau du nerf olfactif, ce qui ouvrirait des possibilités de tests sur des sujets vivants.
La recherche s'intéresse aussi au rôle joué par le système immunitaire pendant la période d'incubation de la maladie. Dans ce domaine, l'équipe de Pierre Aucouturier (Inserm) a montré que des cellules d'un type bien particulier (les cellules dendritiques) peuvent être suffisantes pour transmettre l'infection jusqu'au cerveau, lorsqu'elles sont injectées dans le sang. Si ce résultat laisse encore beaucoup de questions en suspens, il établit une liaison entre les prions et le système immunitaire, qui n'avait encore jamais été explorée.
Plus de risques de contamination chez les jeunes ?
L’une des particularités du nouveau variant de la maladie de Creutzfelt-Jakob par rapport à sa forme classique est l’âge des personnes touchées. La moyenne d’âge est inférieure à 30 ans, alors que la forme classique ne frappe presque jamais avant 60 ans.
De son étude de 2001, l'équipe dirigée par Alain-Jacques Valleron avait déduit que la sensibilité à l'absorption de nourriture contaminée est beaucoup plus grande avant l'âge de 15 ans et qu'elle décroît ensuite très rapidement en fonction de l'âge du consommateur. Ce résultat est cohérent avec l’étude détaillée qui a été menée sur le ''cluster'' de Quenniborough et aucun élément n’est venu le contredire.
Le « cluster » de Queniborough

Queniborough est un petit village d'environ 2000 habitants, situé dans le Leicestershire, au centre de l'Angleterre. Sa particularité est d’avoir enregistré 5 cas de variante humaine de maladie de la vache folle. Les victimes sont décédées entre août 1998 et octobre 2000. Elles avaient entre 19 et 35 ans. Un pourcentage aussi important de personnes atteintes dans un si petit espace ne pouvait qu’attirer l’attention des pouvoirs publics et des scientifiques. En effet, si tous les citoyens du Royaume-Uni avaient été frappés dans les mêmes proportions, c’est près de 150 000 cas qui y seraient apparus.
Une commission de spécialistes a donc été nommée, pour essayer de comprendre comment s’était créé ce “cluster” (échantillon) et tirer des enseignements de ces contaminations concentrées sur un espace géographique très restreint. Après huit mois d'enquête, les docteurs Gerry Bryant et Philip Monk ont rendu leurs conclusions en mars 2001. Leurs investigations les ont dirigés vers deux bouchers dont les pratiques professionnelles traditionnelles (parfaitement autorisées à l’époque) facilitaient la contamination de différents morceaux de viande, y compris la viande rouge, par des matières à risque (tissus nerveux, cerveau, moelle épinière).
Cette étude a permis de justifier – principalement a posteriori – certaines mesures de protection de la santé publique, notamment le retrait des abats à risque et le changement des méthodes d’abattage. Toutefois, les techniques utilisées par ces bouchers détaillants n’expliquent pas l’ensemble des contaminations croisées qui ont pu se produire dans d’autres régions, nombre d’entre elles étant plutôt imputables à des filières industrielles : séparation mécanique de la viande et mélange de tissus nerveux dans les steaks hachés et saucisses, par exemple. Un autre apport de cette étude est d’avoir obtenu des informations sur la durée d'incubation de la maladie, qui a été évaluée entre dix et seize ans pour ces cinq malades des environs de Queniborough.
Toutefois, ce renseignement ne permet aucune conclusion sur la période d’incubation que pourraient connaître d’autres personnes contaminées. Il laisse supposer que la maladie a très peu de chances de se déclencher en un laps de temps plus court mais ne fournit aucun indice sur la durée maximale de cette période, pendant laquelle les patients contaminés ne présentent aucun symptôme.
Une possible protection génétique ?
L'observation des patients déjà atteints a permis de mettre en évidence qu’ils partageaient tous une même particularité génétique, située sur le ''codon 129'', qui n'est présente que chez 40% de la population. On serait alors tenté de penser que les 60% restant seraient protégés contre la maladie. Cette éventualité n’est pourtant pas une certitude et pourrait être remise en question si l'on établit un parallèle avec les cas de maladie de Creutzfeldt-Jakob apparus suite au traitement d'enfants par hormone de croissance. Dans cet exemple, les premiers malades partageaient aussi cette même particularité génétique sur le ''codon 129''.
Mais au cours des années suivantes, des personnes n'ayant pas ce profil ont déclaré la maladie, après un temps d’incubation plus long. La “protection génétique” n’était donc qu’un sursis. Bien évidemment, cette comparaison n'a qu'une valeur indicative car les différences entre les deux cas de figure sont notables. Notamment, pour l'hormone de croissance, les tissus infectieux avaient une origine humaine et aucune barrière d'espèce ne pouvait venir freiner le processus de contamination.
Le codon 129
Le gène qui permet à l’organisme de fabriquer la protéine prion (très répandue sous sa forme normale) se trouve sur le chromosome 20. Une position bien précise de ce gène, appelée “codon 129“, est constituée de deux “bases“ qui peuvent être la méthionine ou la valine.
Il y a donc plusieurs possibilités
- Si ces deux bases sont identiques (deux méthionines ou deux valines), l’individu est dit homozygote pour la méthionine ou la valine.
- Si ces deux bases sont différentes (une méthionine et une valine), l’individu est hétérozygote.
Les homozygotes pour la méthionine (Met-Met sur le codon 129) représentent environ 30 à 40% de la population mais toutes les personnes ayant développé la nouvelle variante de la maladie de Creutzfeldt-Jakob font partie de ce groupe. Il est donc suspecté qu’un lien existe entre cette particularité génétique et la susceptibilité à la forme humaine de la maladie de la vache folle. Pour autant, cette hypothèse n’est pas prouvée et il se pourrait aussi que les personnes homozygotes pour la valine ou hétérozygotes développent un jour la maladie après un temps d’incubation plus long.
Différentes autres mutations sur le gène de la protéine prion ont été observées et reliées à différentes formes d’apparition des maladies à prion (maladie de Creutzfeldt-Jakob sporadique ou insomnie familiale fatale par exemple). Si celle qui concerne le codon 129 est la plus couramment citée, des travaux scientifiques portent sur d’autres sites de ce gène : les codons 102, 117, 178, 200 ainsi que 51 et 118.
Et en France ?
Bien évidemment, toutes les études citées plus haut sont consacrées au Royaume-Uni. Aucun équivalent n'a été établi pour la France. Si notre pays est placé au deuxième rang des nations les plus contaminées, la différence avec la Grande-Bretagne est tellement énorme qu'elle interdit toute comparaison ou extrapolation.
Les neuf cas aujourd'hui recensés dans l'Hexagone ne peuvent fournir aucune base tangible à des travaux statistiques. Il n'est pas non plus possible de tirer d'informations du cas apparu en septembre 2002 en Italie ou de quelques autres cas suspects recensés en Europe et aux Etats-Unis.
