Nobel de chimie 2013 : la modélisation à l'honneur
Annoncé le 9 octobre, le prix Nobel de chimie 2013 a été décerné à Martin Karplus, Michael Levitt et Arieh Warshel, trois pionniers de la modélisation des réactions chimiques qui ont réconcilié la physique newtonnienne et la physique quantique.
Olivier Boulanger - Publié le

En cette fin d'année 2013, pas moins de trois chercheurs se sont vu remettre le prix Nobel de chimie : l’Austro-américain Martin Karplus (université de Strasbourg, France et de Harvard, États-Unis), l’Anglo-américain Michael Levitt (université Stanford), et l’Israélo-américain Arieh Warshel (université de Californie du Sud). Avec ce prix, l’Académie royale suédoise des sciences entend récompenser des travaux décisifs sur la modélisation des réactions chimiques, en l’occurrence sur « le développement de modèles multi-échelle pour les systèmes chimiques complexes »…
Le temps, en effet, où les chimistes utilisaient des boules en plastique et des bâtons pour modéliser les molécules et les réactions chimiques est révolu. Avec le développement, dans les années 1970, des premiers modèles informatiques, les chercheurs disposent aujourd’hui d’outils puissants capables de simuler avec précision des réactions très complexes… Au point que la plupart des chimistes passent désormais plus de temps derrière leurs écrans que devant des éprouvettes. Or, les trois lauréats du Nobel 2013 ne sont pas étrangers à ce changement d’approche.
Réactions virtuelles

Modéliser les réactions chimiques, c’est tenter de reproduire virtuellement les interactions entre les molécules telles que les mouvements d’électrons, les changements de configuration des molécules… Initialement, deux types de modèles s’offraient aux chimistes : ceux s’appuyant sur la physique classique dite « newtonienne », intuitivement la plus simple ; et ceux répondant à la physique quantique, plus précise, mais aussi plus complexe.
L’approche newtonienne permet de modéliser sans problème la structure des grosses molécules. En revanche, et contrairement à l’approche quantique, elle ne permet pas de simuler les réactions chimiques. Les modélisations quantiques sont donc plus performantes, mais elles nécessitent des puissances de calcul telles qu’elles ne peuvent être appliquées qu’à des réactions simples.
Le meilleur des deux mondes

Au début des années 1970, dans son laboratoire d’Harvard, à Cambridge (États-Unis), Martin Karplus travaille justement sur la mise au point de l’un de ces modèles quantiques. Il est rejoint à la même époque par Arieh Warshel qui, en Israël, vient de mettre au point avec Michael Levitt un modèle s’appuyant sur la physique classique.
La collaboration entre Karplus et Warshel aboutit, en 1972, à un programme de modélisation utilisant le « meilleur des deux mondes ». Appliqué à une molécule proche du « rétinal » – la molécule de la vision –, ce programme recourt à un modèle quantique pour simuler le mouvement des électrons libres et relègue à la physique classique le reste de la modélisation. Une première ! même si ce modèle inédit présente encore de nombreuses limitations.
Une approche toujours d'actualité

L’objectif à terme, néanmoins, reste de pouvoir simuler n’importe quelle réaction, et notamment les réactions enzymatiques, primordiales pour comprendre la chimie du vivant. Dès 1976, en collaboration avec Michael Levitt, un nouveau modèle révolutionnaire est ainsi réalisé, le tout premier à pouvoir simuler le fonctionnement d’une enzyme. En outre, celui-ci peut aussi s’appliquer à d’autres molécules complexes.
Aujourd’hui encore, la modélisation des réactions chimiques ne diffère guère du principe élaboré par les trois chercheurs dans les années 1970. Le centre de la réaction chimique, mettant en œuvre des interactions entre électrons et noyaux atomiques, est pris en charge par un modèle quantique ; le reste, par des équations relevant de la physique classique.
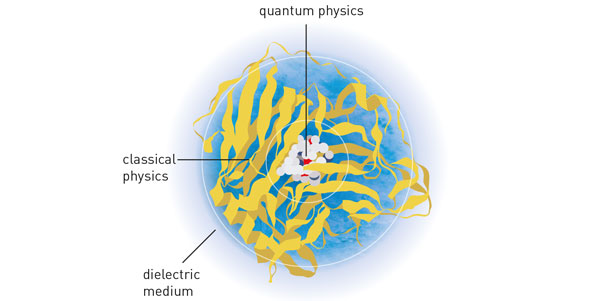
Une troisième couche de simulation peut éventuellement être ajoutée au modèle : dans les régions éloignées de la réaction chimique, certains groupements d’atomes peuvent être assimilés à des masses homogènes – toujours dans un souci de simplification – sans conséquence sur le résultat de la réaction chimique.
Le rêve des chercheurs ? Michael Levitt l’exprime dans l’une de ces publications : simuler un organisme vivant à l’échelle moléculaire. Un long chemin reste à faire pour y parvenir. Mais qui sait ? La puissance des calculateurs étant exponentielle, ce rêve pourrait bien devenir réalité dans les années à venir.
