Thérapie génique : un premier succès pour la bêta-thalassémie
Un jeune homme de 18 ans atteint d'une forme grave de thalassémie, une maladie du sang qui touche 5% de la population mondiale, s'est porté volontaire en 2007 pour un essai de thérapie génique. Il est aujourd'hui en bonne santé et mène une vie normale...
Paloma Bertrand - Publié le
Une maladie génétique fréquente
Les hémoglobinopathies (bêta-thalassémie et drépanocytose) sont les maladies génétiques les plus fréquentes au monde. Près de 5% de la population mondiale, principalement sur le pourtour méditerranéen, le Moyen-Orient, l'Afrique sub-saharienne et l'Asie est porteuse d'un gène de globine défectueux et 300 000 enfants naissent chaque année avec une forme grave d'hémoglobinopathie.
La bêta-thalassémie est due à un défaut du gène codant pour la bêta-globine, un constituant essentiel de l'hémoglobine. Présente dans les globules rouges, cette molécule transporte l'oxygène depuis les poumons vers tous les tissus de l'organisme. Dans les formes « légères », les patients peuvent souffrir d'anémie modérée mais dans les cas les plus graves, le manque d'oxygène dans le sang est mortel : sans traitement, l'espérance de vie de ces malades est d'environ 8 ans.
Des traitements lourds
Le traitement conventionnel de la thalassémie, pour les formes les plus graves, est basé sur des transfusions sanguines, répétées toutes les 3 à 4 semaines, dès que le taux d'hémoglobine descend en dessous d'une valeur compatible avec une activité normale. Mais la répétition de ces transfusions conduit à un excès de fer qui doit être éliminé par un traitement approprié, afin d'éviter des effets secondaires graves et parfois mortels. Si ce traitement a transformé l'espérance de vie des patients, sa lourdeur altère fortement leur qualité de vie.
Quand c'est possible, une greffe de moelle osseuse peut être envisagée chez les enfants et les adolescents, mais encore faut-il trouver un donneur compatible. De plus, il y a toujours un risque de rejet de la greffe. La mise au point d'une thérapie génique est donc un espoir pour les 80 % de malades ne pouvant pas avoir accès à une greffe de moelle.
Apport d’un gène par un lentivirus dérivé du VIH
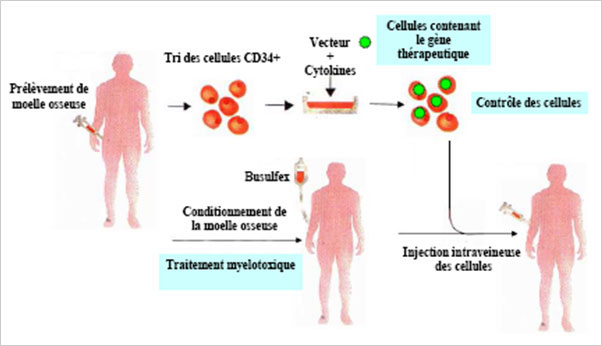
L'essai conduit par l'équipe franco-américaine dirigée par le professeur Philippe Leboulch (Inserm, CEA, université Paris-Sud) a été lancé en France, en 2007, sur un jeune homme âgé à l'époque de 18 ans et dépendant depuis son enfance de transfusions mensuelles. Le traitement a débuté par un prélèvement de cellules de moelle osseuse du patient dont on a extrait les cellules souches du sang, porteuses du gène défectueux. Ces cellules ont ensuite été mises en contact avec un lentivirus (un virus dérivé du VIH mais rendu inoffensif) porteur d'une version fonctionnelle du gène. Ce lentivirus, capable de pénétrer dans le noyau des cellules, est allé s'intégrer dans le génome avec le gène opérationnel. Le patient a alors reçu une chimiothérapie pour détruire ses propres cellules de moelle osseuse défectueuses. Et 48 heures après, les cellules souches modifiées lui ont été injectées par voie intraveineuse.
11 mois plus tard, le patient n'avait plus besoin de transfusion. « L'insertion par thérapie génique d'une copie du gène de la bêta-globine a permis, au bout de quelques mois, aux cellules d'exprimer la bêta-globine à un taux suffisamment élevé pour que le patient n'ait plus besoin de transfusion, et ce depuis plus de deux ans ! C'est une avancée historique dans le combat contre cette maladie », témoigne le Pr. Marina Cavazzana-Calvo, directrice du département de biothérapie de l'hôpital Necker-Enfants malades. Suite à ce résultat prometteur, l'Afssaps a autorisé l'inclusion d'un nouveau patient dans ce protocole de thérapie génique. En parallèle, un accord de collaboration a été signé avec la Thaïlande pour étendre l'essai clinique à ce pays où la bêta-thalassémie est un problème de santé publique, avec plus de 3 000 naissances par an d'enfants atteints de cette maladie.
